Denaturing and temperature gradient
gel electrophoresis (DGGE and TGGE) are methods of generating
fingerprints of microbial communities. DGGE
was first described by in
1979, but
wasn’t applied to profiling microbial communities until 1993.
How does DGGE work?
The technique takes advantage of the different melting domains of double stranded DNA based on sequence and that a partially melted DNA molecule will stop moving through an agarose gel allowing for separation of sequences based on a difference of one base pair. A gradient of either temperature or a denaturant such as formamide or urea is created over an agarose gel. When a sample of different 16S rRNA sequences is run through the gel, they will separate according to differences in nucleotide composition (and therefore phylogeny) rather than size. In general, the higher the G-C content of a sequence, the further through the gel it will advance.
Each microbial community will produce a different pattern of bands depending on its composition so the technique can be applied to investigate differences in genetic diversity between microbial communities or in the same community over time. DGGE will detect microorganisms which contribute to more than 1% of the total microbial population. If we want to examine less abundant groups, we could do a PCR on our original samples using group specific primers.
How does DGGE work?
The technique takes advantage of the different melting domains of double stranded DNA based on sequence and that a partially melted DNA molecule will stop moving through an agarose gel allowing for separation of sequences based on a difference of one base pair. A gradient of either temperature or a denaturant such as formamide or urea is created over an agarose gel. When a sample of different 16S rRNA sequences is run through the gel, they will separate according to differences in nucleotide composition (and therefore phylogeny) rather than size. In general, the higher the G-C content of a sequence, the further through the gel it will advance.
Each microbial community will produce a different pattern of bands depending on its composition so the technique can be applied to investigate differences in genetic diversity between microbial communities or in the same community over time. DGGE will detect microorganisms which contribute to more than 1% of the total microbial population. If we want to examine less abundant groups, we could do a PCR on our original samples using group specific primers.

After electrophoresis has separated different sequences, they can be cut out and sequence the DNA that's creating the band. If DGGE is going to be used to analyse the products of PCR amplification, primers with a GC-clamp (a 40 base pair sequence composed entirely of GC base pairs) are required. The GC-clamp prevents complete separation of DNA strands to form single stranded DNA which would continue to advance through the agarose gel and form extra bands.
What you get out the other end is something that looks like this:
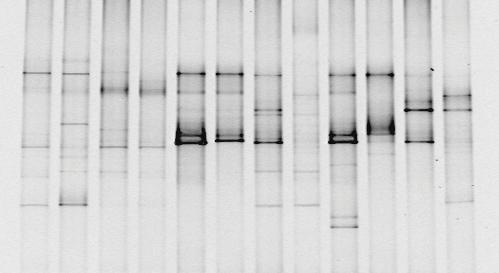 |
| Bioguz at English Wikipedia, 16S PCR DGGE, CC BY-SA 3.0 |
{kind=link}
Biases and Limitations
As with any technique there are going to be some limitations and other factors to take into account when we're analysing the results. At this stage, those pesky heteroduplexes and chimeras from the PCR are going to start causing problems.
Diversity may be overestimated because of:
- Chimeras – formed during PCR and will behave as a normal rDNA sequence. Removal and sequencing of a band could allow for detection of chimeric sequences with the relevant software.
- Intraspecific rDNA heterogeneities – Since single species may have rRNA operons which differ by as much as 1%, these can form separate bands in the gel.
- Heterduplexes – Since heteroduplexes are less stable and they can appear as unspecific bands within the gel. However, it is generally thought that heteroduplexes do not interfere with DGGE analysis.
It's also possible that we might lose some diversity because of:
- Comigration of fragments – As the separation of fragments if based on G-C content and not nucleotide sequence, it is possible that fragments with a different sequence will migrate to the same position in the gel.
- Merging of bands – Two rDNA fragments with similar sequences can migrate to adjacent positions on the gel. If the bands are close enough, they may merge and appear as a single band.
As well as these biases, DGGE has some limitations to the technique:
- Small fragment size – While DGGE allows for excision and sequencing of bands, rDNA fragments are only 500 basepairs. This provides limited information for phylogenetic anaylsis. This can be overcome by relying co-analysis with clone libraries containing longer rDNA fragments which can then be used to construct phylogenetic trees.
- Gel-to-gel variation – Subtle differences in gel preparation limit the viability of comparing samples run on different gels. This limits the number of samples that can be compared as they must all be run on the same gel. Software is now available to help minimise the effect of gel-to-gel variation.
- DGGE is semi-quantitative – As we mentioned before when talking about differential PCR amplification, some bacteria have more than one copy of the 16S rRNA gene. If the different DNA fragments migrate to the same point on the gel, this will give an inaccurate intensity of that band. Equally, different DNA fragments from the same species can migrate to form different bands on the gel, giving a false impression of diversity.
No comments:
Post a Comment